
Kod
library(tidymodels)
tidymodels_prefer()
library(tidyverse)
library(beans)Redukcja wymiarowości (ang. dimensionality reduction) jest przekształceniem zbioru danych z przestrzeni wielowymiarowej w przestrzeń niskowymiarową i może być dobrym wyborem, gdy podejrzewamy, że jest “za dużo” zmiennych. Nadmiar zmiennych, zwykle predyktorów, może być problemem, ponieważ trudno jest zrozumieć lub wizualizować dane w wyższych wymiarach.
Redukcja wymiarowości może być stosowana zarówno w inżynierii cech, jak i w eksploracyjnej analizie danych. Na przykład, w wielowymiarowych eksperymentach biologicznych, jednym z pierwszych zadań, przed jakimkolwiek modelowaniem, jest określenie, czy istnieją jakiekolwiek niepożądane trendy w danych (np. efekty niezwiązane z interesującym nas zagadnieniem, takie jak różnice między laboratoriami). Eksploracja danych jest trudna, gdy istnieją setki tysięcy wymiarów, a redukcja wymiarowości może być pomocą w analizie danych.
Inną potencjalną konsekwencją posiadania wielu predyktorów jest model nadmiarową liczbą predyktorów. Najprostszym przykładem jest regresja liniowa, gdzie liczba predyktorów powinna być mniejsza niż liczba obserwacji użytych do dopasowania modelu. Innym problemem jest współliniowość, gdzie korelacje między predyktorami mogą negatywnie wpływać na operacje matematyczne używane do oszacowania modelu. Jeśli istnieje bardzo duża liczba predyktorów, jest mało prawdopodobne, że istnieje taka sama liczba rzeczywistych efektów leżących u podstaw. Predyktory mogą mierzyć ten sam ukryty efekt (efekty), a zatem takie predyktory będą wysoko skorelowane. Wiele technik redukcji wymiarowości sprawdza się w tej sytuacji. W rzeczywistości większość z nich może być skuteczna tylko wtedy, gdy istnieją takie relacje między predyktorami, które można wykorzystać.
Analiza składowych głównych (PCA) jest jedną z najprostszych metod redukcji liczby zmiennych w zbiorze danych, ponieważ opiera się na metodach liniowych i jest nienadzorowana. W przypadku wielowymiarowego problemu klasyfikacji, początkowy wykres głównych komponentów PCA może pokazać wyraźny podział między klasami. Jeśli tak jest, to można bezpiecznie założyć, że klasyfikator liniowy może się sprawdzić w takim zadaniu. Jednak nie jest prawdą, że brak separacji nie oznacza, że problem jest nie do pokonania.
Metody redukcji wymiarowości omawiane w tym rozdziale nie są na ogół metodami selekcji cech. Metody takie jak PCA reprezentują oryginalne predyktory za pomocą mniejszego podzbioru nowych cech. Wszystkie oryginalne predyktory są wymagane do obliczenia tych nowych cech. Wyjątkiem są metody rzadkie, które mają zdolność do całkowitego usunięcia wpływu predyktorów podczas tworzenia nowych cech.
Do celów ilustracji, jak używać redukcji wymiarowości z przepisami wykorzystamy przykładowy zestaw danych. Koklu i Ozkan (2020) opublikowali zestaw danych dotyczących wizualnych cech suszonej fasoli i opisali metody określania odmian suszonej fasoli na obrazie. Chociaż wymiarowość tych danych nie jest bardzo duża w porównaniu z wieloma problemami modelowania w świecie rzeczywistym, zapewnia ładny przykład roboczy, aby zademonstrować, jak zmniejszyć liczbę cech. W swojej pracy napisali:
Podstawowym celem niniejszej pracy jest dostarczenie metody uzyskiwania jednolitych odmian nasion z produkcji roślinnej, która ma postać populacji, więc nasiona nie są certyfikowane jako jedyna odmiana. W związku z tym opracowano system wizji komputerowej do rozróżniania siedmiu różnych zarejestrowanych odmian suchej fasoli o podobnych cechach w celu uzyskania jednolitej klasyfikacji nasion. Dla modelu klasyfikacji wykonano obrazy 13 611 ziaren 7 różnych zarejestrowanych suchych odmian fasoli za pomocą kamery o wysokiej rozdzielczości.
Każdy obraz zawiera wiele ziaren. Proces określania, które piksele odpowiadają konkretnej fasoli, nazywany jest segmentacją obrazu. Te piksele mogą być analizowane w celu uzyskania cech dla każdej fasoli, takich jak kolor i morfologia (tj. kształt). Cechy te są następnie wykorzystywane do modelowania wyniku (odmiany fasoli), ponieważ różne odmiany fasoli wyglądają inaczej. Dane treningowe pochodzą z zestawu ręcznie oznakowanych obrazów, a ten zestaw danych jest używany do tworzenia modelu predykcyjnego, który może rozróżnić siedem odmian fasoli: Cali, Horoz, Dermason, Seker, Bombay, Barbunya i Sira. Stworzenie skutecznego modelu może pomóc producentom w ilościowym określeniu jednorodności partii fasoli.
Istnieje wiele metod kwantyfikacji kształtów obiektów (Mingqiang, Kidiyo, i Joseph 2008). Wiele z nich dotyczy granic lub regionów interesującego nas obiektu. Przykładowe cechy obejmują:
W danych dotyczących fasoli obliczono 16 cech morfologicznych: powierzchnię, obwód, długość osi głównej, długość osi małej, współczynnik kształtu, ekscentryczność, powierzchnię wypukłą, średnicę równoważną, rozległość, zwartość, krągłość, zwięzłość, współczynnik kształtu 1, współczynnik kształtu 2, współczynnik kształtu 3 i współczynnik kształtu 4.
library(tidymodels)
tidymodels_prefer()
library(tidyverse)
library(beans)Dla naszych analiz zaczynamy od podziału zbioru za pomocą initial_split(). Pozostałe dane są dzielone na zbiory treningowe i walidacyjne:
<Training/Validation/Total>
<8163/2043/10206>Aby wizualnie ocenić, jak dobrze działają różne metody, możemy oszacować metody na zbiorze treningowym (n = 8163 ziaren) i wyświetlić wyniki przy użyciu zbioru walidacyjnego (n = 2043).
Przed rozpoczęciem jakiejkolwiek redukcji wymiarowości możemy poświęcić trochę czasu na zbadanie naszych danych. Ponieważ wiemy, że wiele z tych cech kształtu prawdopodobnie mierzy podobne koncepcje, przyjrzyjmy się strukturze korelacyjnej danych na Rys. 14.1.
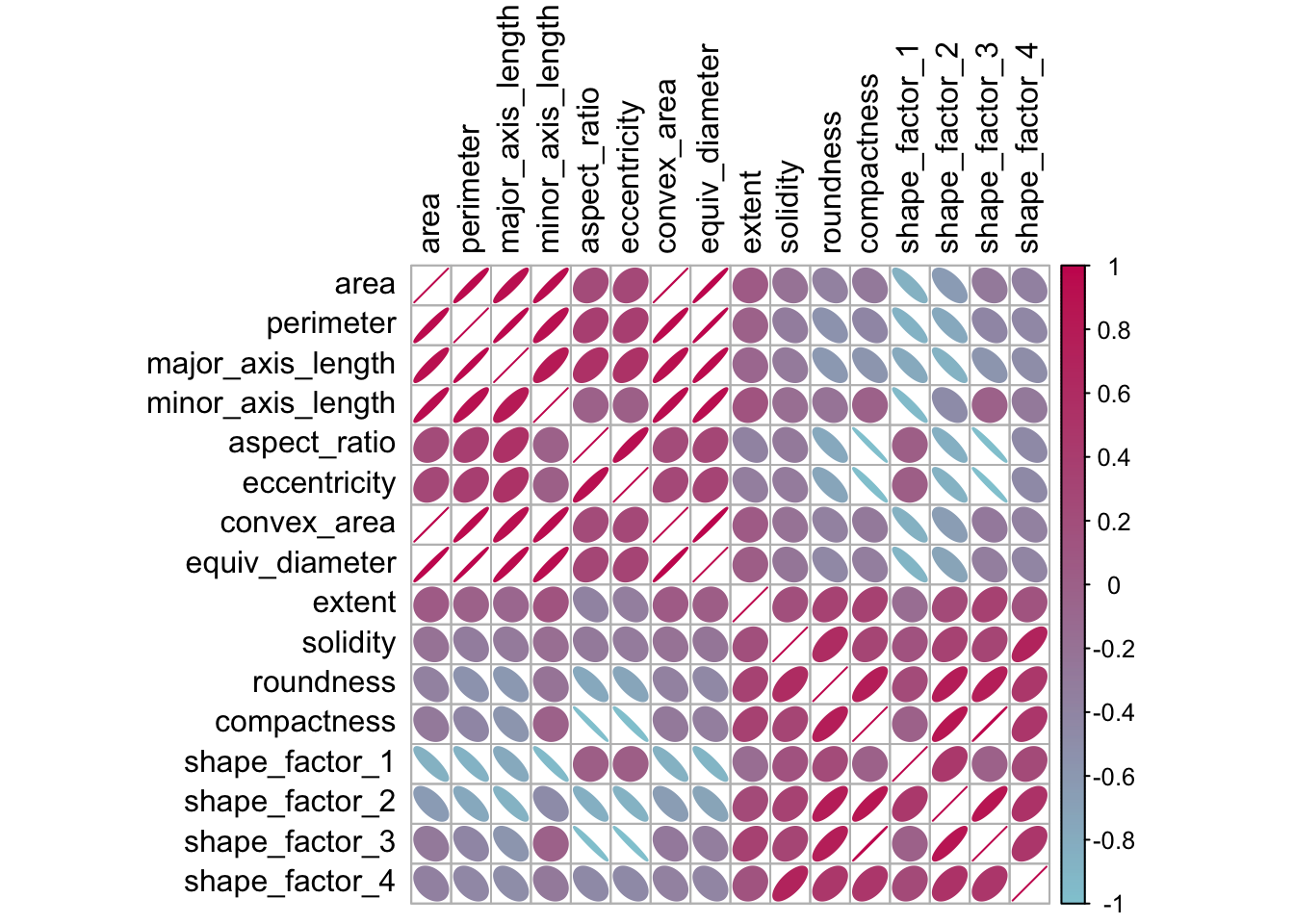
Wiele z tych predyktorów jest silnie skorelowanych, jak np. powierzchnia i obwód lub współczynniki kształtu 2 i 3. Chociaż nie poświęcamy temu czasu tutaj, ważne jest również, aby zobaczyć, czy ta struktura korelacji znacząco zmienia się w różnych kategoriach wyników. Może to pomóc w stworzeniu lepszych modeli.
Zacznijmy od podstawowego przepisu wstępnego przetwarzania danych, który często stosujemy przed jakimikolwiek krokami redukcji wymiarowości. Kilka predyktorów to współczynniki, a więc prawdopodobnie będą miały skośne rozkłady. Takie rozkłady mogą siać spustoszenie w obliczeniach wariancji (takich jak te używane w PCA). Pakiet bestNormalize posiada krok, który może wymusić symetryczny rozkład predyktorów. Użyjemy tego, aby złagodzić problem skośnych rozkładów:
library(bestNormalize)
bean_rec <-
# Use the training data from the bean_val split object
recipe(class ~ ., data = analysis(bean_val$splits[[1]])) %>%
step_zv(all_numeric_predictors()) %>%
step_orderNorm(all_numeric_predictors()) %>%
step_normalize(all_numeric_predictors())Pamiętaj, że podczas wywoływania funkcji recipe() kroki nie są w żaden sposób szacowane ani wykonywane.
Przepis ten zostanie rozszerzony o dodatkowe kroki dla analiz redukcji wymiarowości. Zanim to zrobimy, przejdźmy do tego, jak receptura może być używana poza przepływem pracy.
Przepływ pracy zawierający recepturę wykorzystuje fit() do estymacji receptury i modelu, a następnie predict() do przetwarzania danych i tworzenia przewidywań modelu. W pakiecie recipes znajdują się analogiczne funkcje, które mogą być użyte do tego samego celu:
prep(recipe, training) dopasowuje przepis do zbioru treningowego.bake(recipe, new_data) stosuje operacje receptury do new_data.Rysunek 16.3 podsumowuje to.

bean_rec_trained <- prep(bean_rec)
bean_rec_trainedZauważ, że kroki zostały wytrenowane i że selektory nie są już ogólne (tj. all_numeric_predictors()); teraz pokazują rzeczywiste kolumny, które zostały wybrane. Również, prep(bean_rec) nie wymaga argumentu training. Możesz przekazać dowolne dane do tego argumentu, ale pominięcie go oznacza, że użyte zostaną oryginalne dane z wywołania recipe(). W naszym przypadku były to dane ze zbioru treningowego.
Jednym z ważnych argumentów funkcji prep() jest retain. Kiedy retain = TRUE (domyślnie), szacunkowa wersja zbioru treningowego jest przechowywana wewnątrz receptury. Ten zestaw danych został wstępnie przetworzony przy użyciu wszystkich kroków wymienionych w recepturze. Ponieważ funkcja prep() musi wykonywać recepturę w trakcie jej wykonywania, korzystne może być zachowanie tej wersji zbioru treningowego, tak że jeśli ten zbiór danych ma być użyty później, można uniknąć zbędnych obliczeń. Jednakże, jeśli zestaw treningowy jest duży, może być problematyczne przechowywanie tak dużej ilości danych w pamięci. Użyj wówczas retain = FALSE, aby tego uniknąć.
Po dodaniu nowych kroków do oszacowanej receptury, ponowne zastosowanie prep() oszacuje tylko niewykształcone kroki. Przyda się to, gdy będziemy próbować różnych metod ekstrakcji cech. Inną opcją, która może pomóc zrozumieć, co dzieje się w analizie, jest log_changes:
show_variables <-
bean_rec %>%
prep(log_changes = TRUE)step_zv (zv_6JtxV): same number of columns
step_orderNorm (orderNorm_4r8al): same number of columns
step_normalize (normalize_x6oqH): same number of columnsNa przykład, próbki zbioru walidacyjnego mogą być przetwarzane następująco:
Rys. 14.3 przedstawia histogramy predyktora powierzchni przed i po przygotowaniu receptury.
library(patchwork)
p1 <-
bean_validation %>%
ggplot(aes(x = area)) +
geom_histogram(bins = 30, color = "white", fill = "blue", alpha = 1/3) +
ggtitle("Original validation set data")
p2 <-
bean_val_processed %>%
ggplot(aes(x = area)) +
geom_histogram(bins = 30, color = "white", fill = "red", alpha = 1/3) +
ggtitle("Processed validation set data")
p1 + p2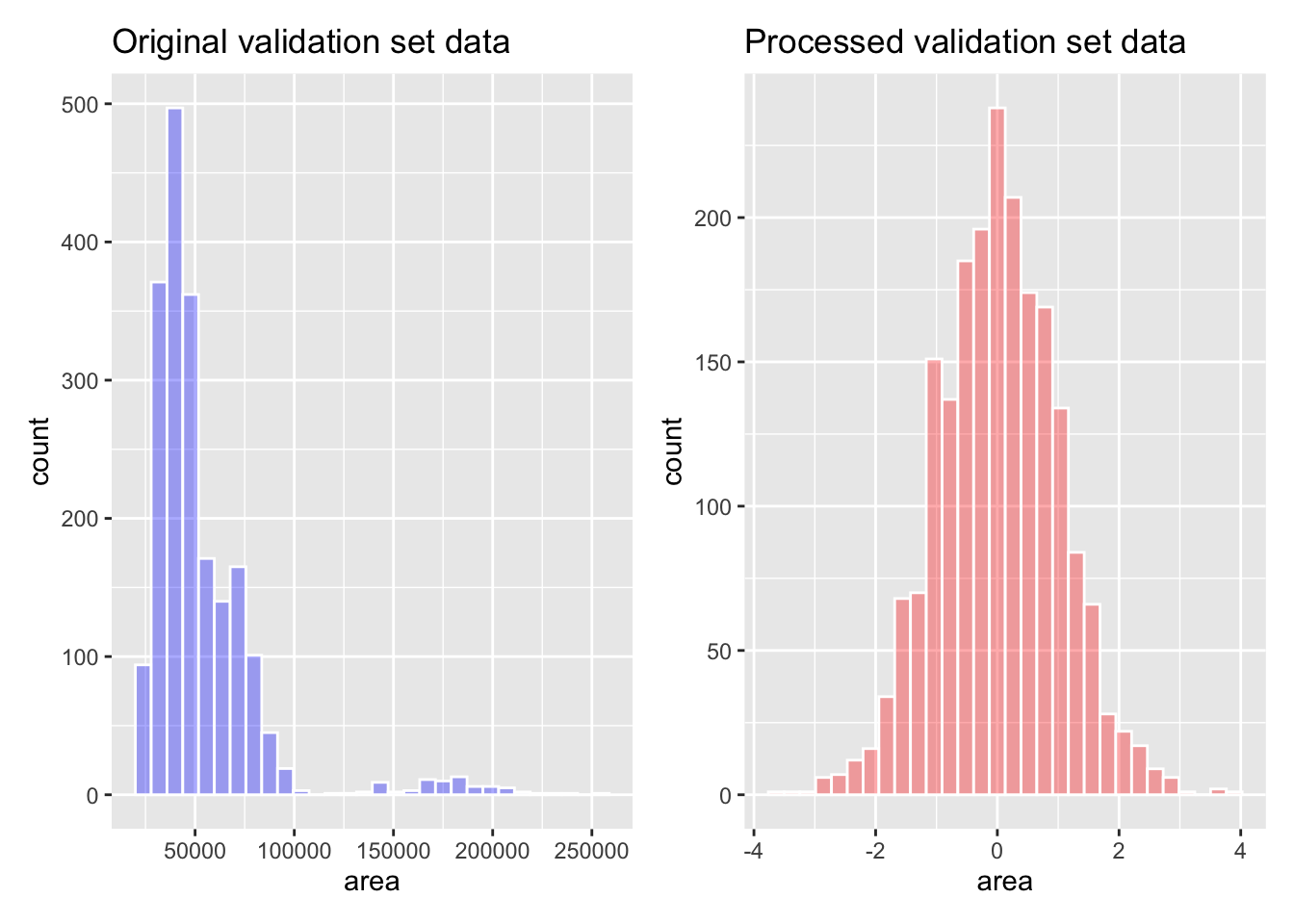
Warto tutaj zwrócić uwagę na dwa ważne aspekty bake(). Po pierwsze, jak wcześniej wspomniano, użycie prep(recipe, retain = TRUE) zachowuje istniejącą przetworzoną wersję zbioru treningowego w recepturze. Dzięki temu użytkownik może użyć bake(recipe, new_data = NULL), która zwraca ten zestaw danych bez dalszych obliczeń. Na przykład:
[1] 8163[1] 8163Jeśli zestaw treningowy nie jest patologicznie duży, użycie retain może zaoszczędzić dużo czasu obliczeniowego. Po drugie, w wywołaniu można użyć dodatkowych selektorów, aby określić, które kolumny mają zostać zwrócone. Domyślnym selektorem jest everything(), ale można dokonać bardziej szczegółowych wyborów.
Ponieważ receptury są podstawową opcją w tidymodels do redukcji wymiarowości, napiszmy funkcję, która oszacuje transformację i wykreśli wynikowe dane w postaci macierzy wykresów za pośrednictwem pakietu ggforce:
library(ggforce)
plot_validation_results <- function(recipe, dat = assessment(bean_val$splits[[1]])) {
recipe %>%
# Estimate any additional steps
prep() %>%
# Process the data (the validation set by default)
bake(new_data = dat) %>%
# Create the scatterplot matrix
ggplot(aes(x = .panel_x, y = .panel_y, color = class, fill = class)) +
geom_point(alpha = 0.4, size = 0.5) +
geom_autodensity(alpha = .3) +
facet_matrix(vars(-class), layer.diag = 2) +
scale_color_brewer(palette = "Dark2") +
scale_fill_brewer(palette = "Dark2")
}Funkcji tej użyjemy wielokrotnie do wizualizacji wyników po zastosowaniu różnych metod ekstrakcji cech.
PCA jest metodą bez nadzoru, która wykorzystuje liniowe kombinacje predyktorów do określenia nowych cech. Cechy te starają się uwzględnić jak najwięcej zmienności w oryginalnych danych. Dodajemy step_pca() do oryginalnego przepisu i wykorzystujemy naszą funkcję do wizualizacji wyników na zbiorze walidacyjnym na Rys. 14.4 za pomocą:
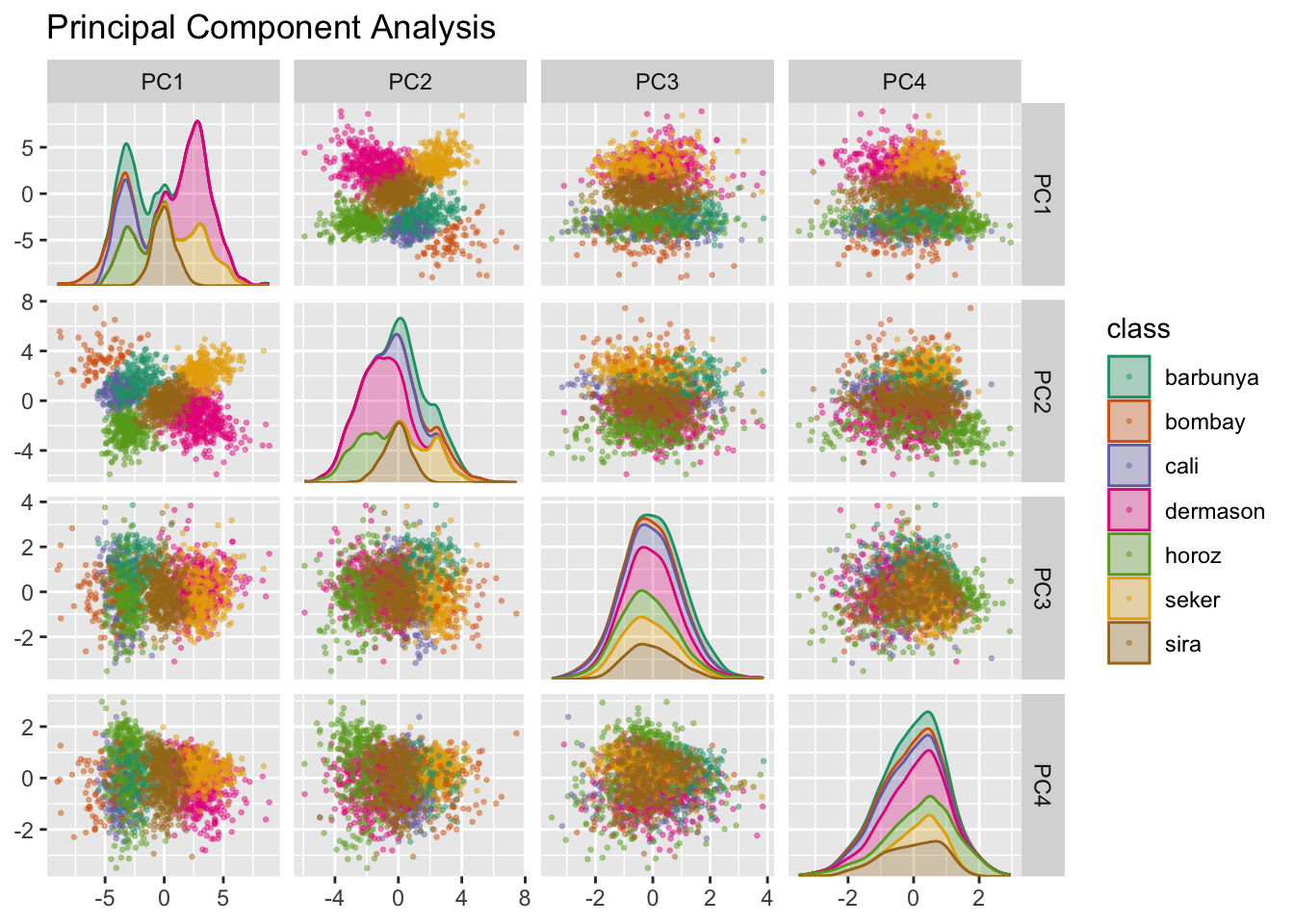
Widzimy, że pierwsze dwie składowe PC1 i PC2, zwłaszcza gdy są używane razem, skutecznie rozróżniają lub oddzielają klasy. To może nas skłonić do oczekiwania, że ogólny problem klasyfikacji tych ziaren nie będzie szczególnie trudny.
Przypomnijmy, że PCA jest metodą bez nadzoru. Dla tych danych okazuje się, że składowe PCA, które wyjaśniają największe różnice w predyktorach, są również predyktorami klas. Jakie cechy wpływają na wydajność? Pakiet learntidymodels posiada funkcje, które mogą pomóc w wizualizacji najważniejszych cech dla każdego komponentu. Będziemy potrzebowali przygotowanego przepisu; krok PCA jest dodany w poniższym kodzie wraz z wywołaniem prep():
library(learntidymodels)
bean_rec_trained %>%
step_pca(all_numeric_predictors(), num_comp = 4) %>%
prep() %>%
plot_top_loadings(component_number <= 4, n = 5) +
scale_fill_brewer(palette = "Paired") +
ggtitle("Principal Component Analysis")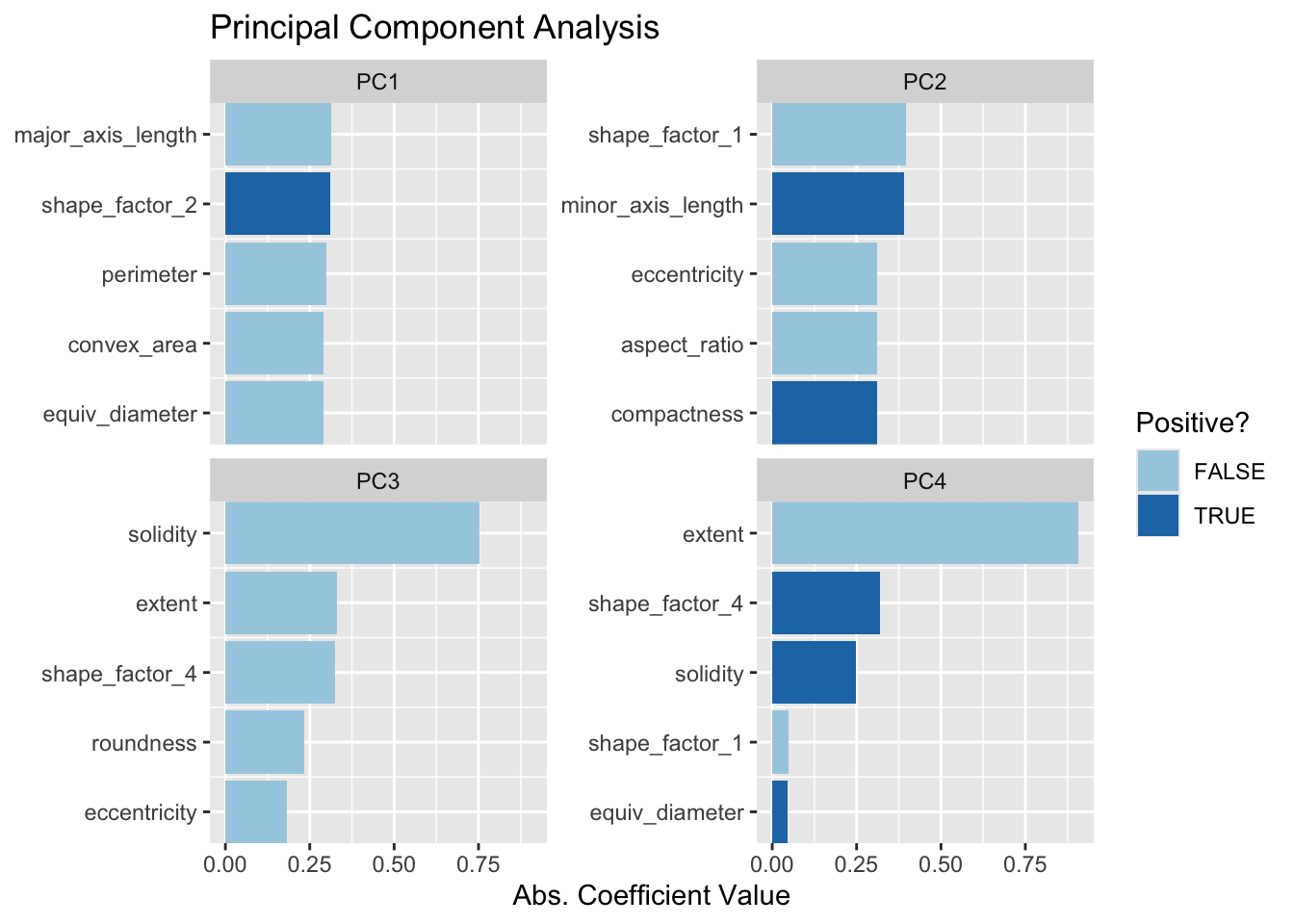
Najwyższe ładunki są w większości związane z grupą skorelowanych predyktorów pokazanych w lewej górnej części poprzedniego wykresu korelacji: obwód, powierzchnia, długość osi głównej i powierzchnia wypukła. Wszystkie one są związane z wielkością fasoli. Miary wydłużenia wydają się dominować w drugim komponencie PCA.
Partial Least Squares jest nadzorowaną wersją PCA. Próbuje znaleźć składowe, które jednocześnie maksymalizują zmienność predyktorów, a jednocześnie maksymalizują związek między tymi składowymi a wynikiem. Rys. 14.6 przedstawia wyniki tej nieco zmodyfikowanej wersji kodu PCA:
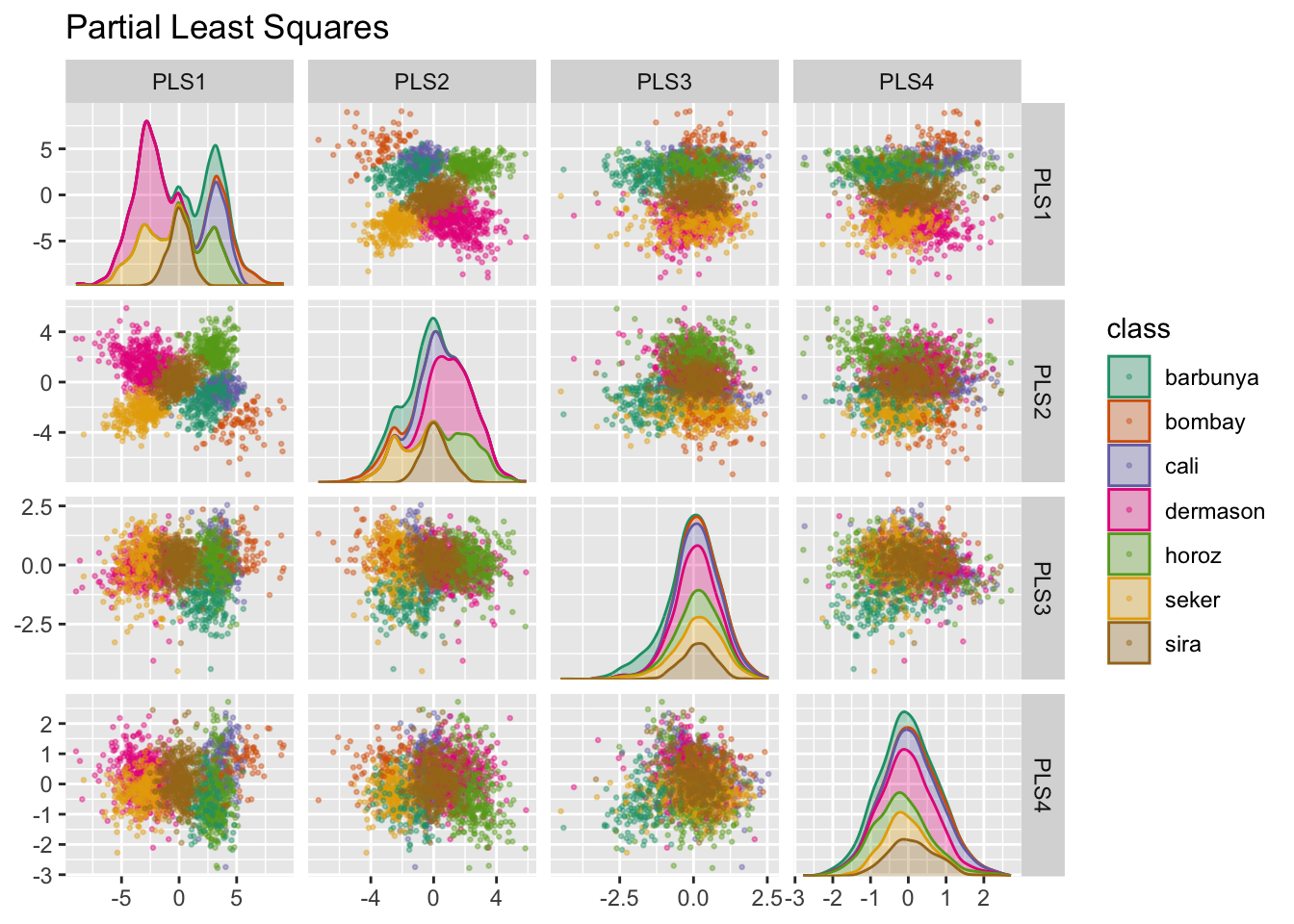
Pierwsze dwie składowe PLS wykreślone na Rys. 14.6 są niemal identyczne z pierwszymi dwiema składowymi PCA! Pozostałe składowe różnią się nieco od PCA. Rys. 14.7 wizualizuje ładunki, czyli najwyższe cechy dla każdej składowej.
bean_rec_trained %>%
step_pls(all_numeric_predictors(), outcome = "class", num_comp = 4) %>%
prep() %>%
plot_top_loadings(component_number <= 4, n = 5, type = "pls") +
scale_fill_brewer(palette = "Paired") +
ggtitle("Partial Least Squares")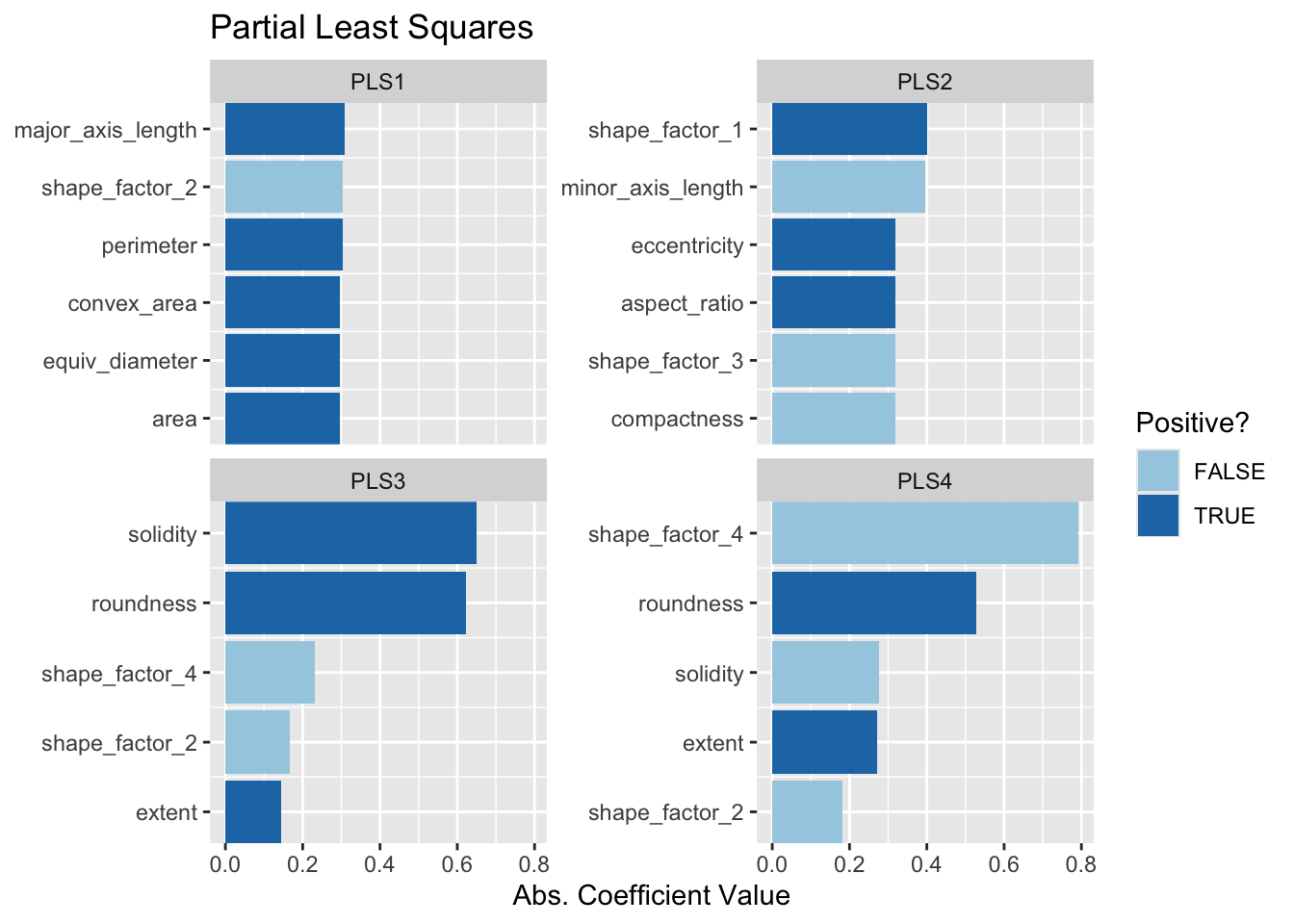
Metoda składowych niezależnych różni się nieco od PCA tym, że znajduje składowe, które są tak statystycznie niezależne od siebie, jak to tylko możliwe (w przeciwieństwie do bycia nieskorelowanym). Można o tym myśleć jako o rozdzielaniu informacji zamiast kompresji informacji jak w przypadku PCA. Użyjmy funkcji step_ica() do wykonania dekompozycji (patrz Rys. 14.8):
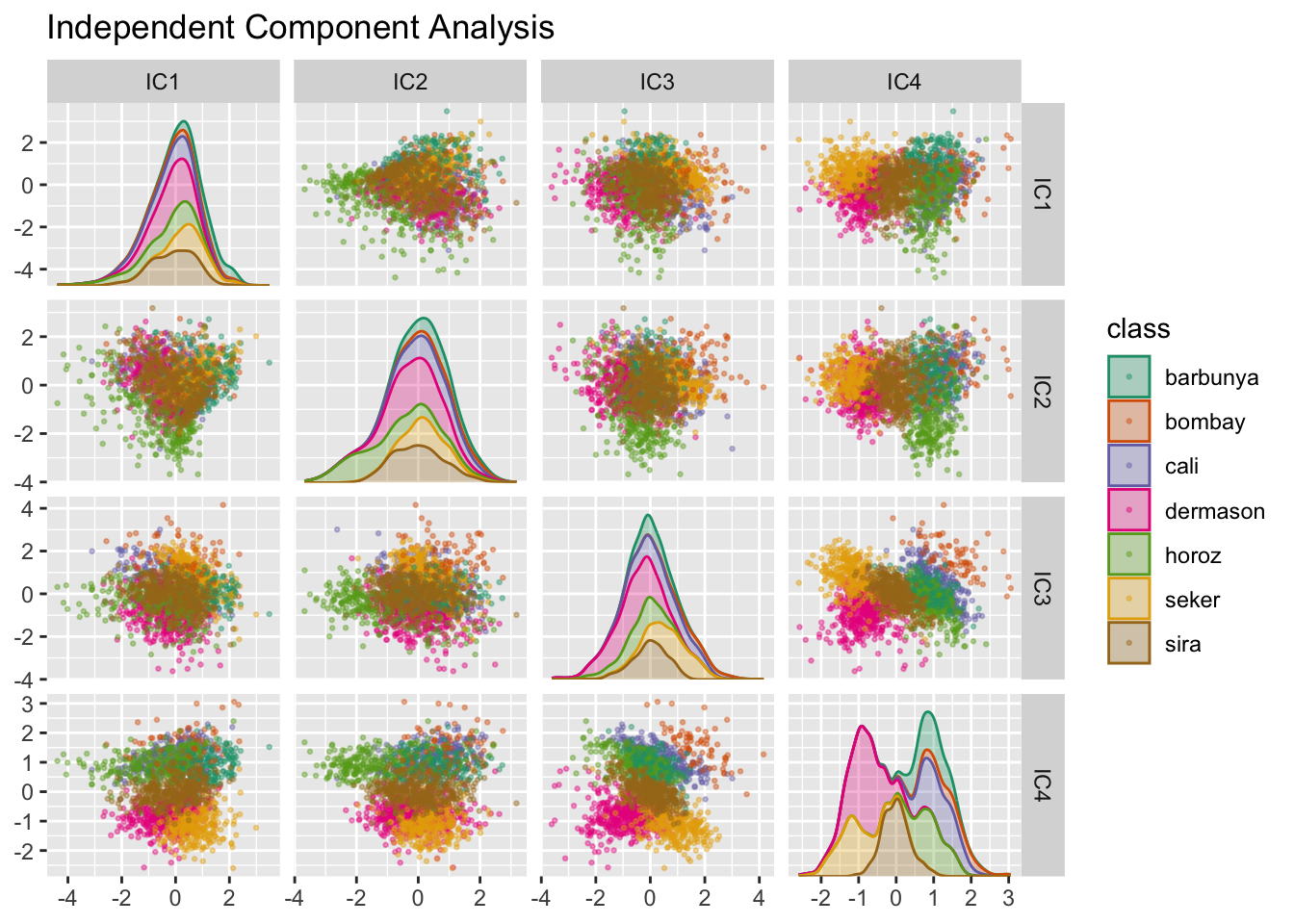
Analiza wyników nie wskazuje na wyraźne różnice między klasami w pierwszych kilku komponentach ICA. Te niezależne (lub tak niezależne, jak to możliwe) komponenty nie separują typów fasoli.
Uniform manifold approximation and projection jest podobna do innej znanej metody t-SNE służącej do nieliniowej redukcji wymiarów. W oryginalnej przestrzeni wielowymiarowej UMAP wykorzystuje opartą na odległości metodę najbliższych sąsiadów, aby znaleźć lokalne obszary danych, w których punkty z dużym prawdopodobieństwem są powiązane. Relacje między punktami danych są zapisywane jako model grafu skierowanego, gdzie większość punktów nie jest połączona. Stąd UMAP tłumaczy punkty w grafie na przestrzeń o zmniejszonym wymiarze. Aby to zrobić, algorytm posiada proces optymalizacji, który wykorzystuje entropię krzyżową do mapowania punktów danych do mniejszego zestawu cech, tak aby graf był dobrze przybliżony.
Pakiet embed zawiera funkcję krokową dla tej metody, zwizualizowaną na Rys. 14.9.
bean_rec_trained %>%
step_umap(all_numeric_predictors(), num_comp = 4) %>%
plot_validation_results() +
ggtitle("UMAP")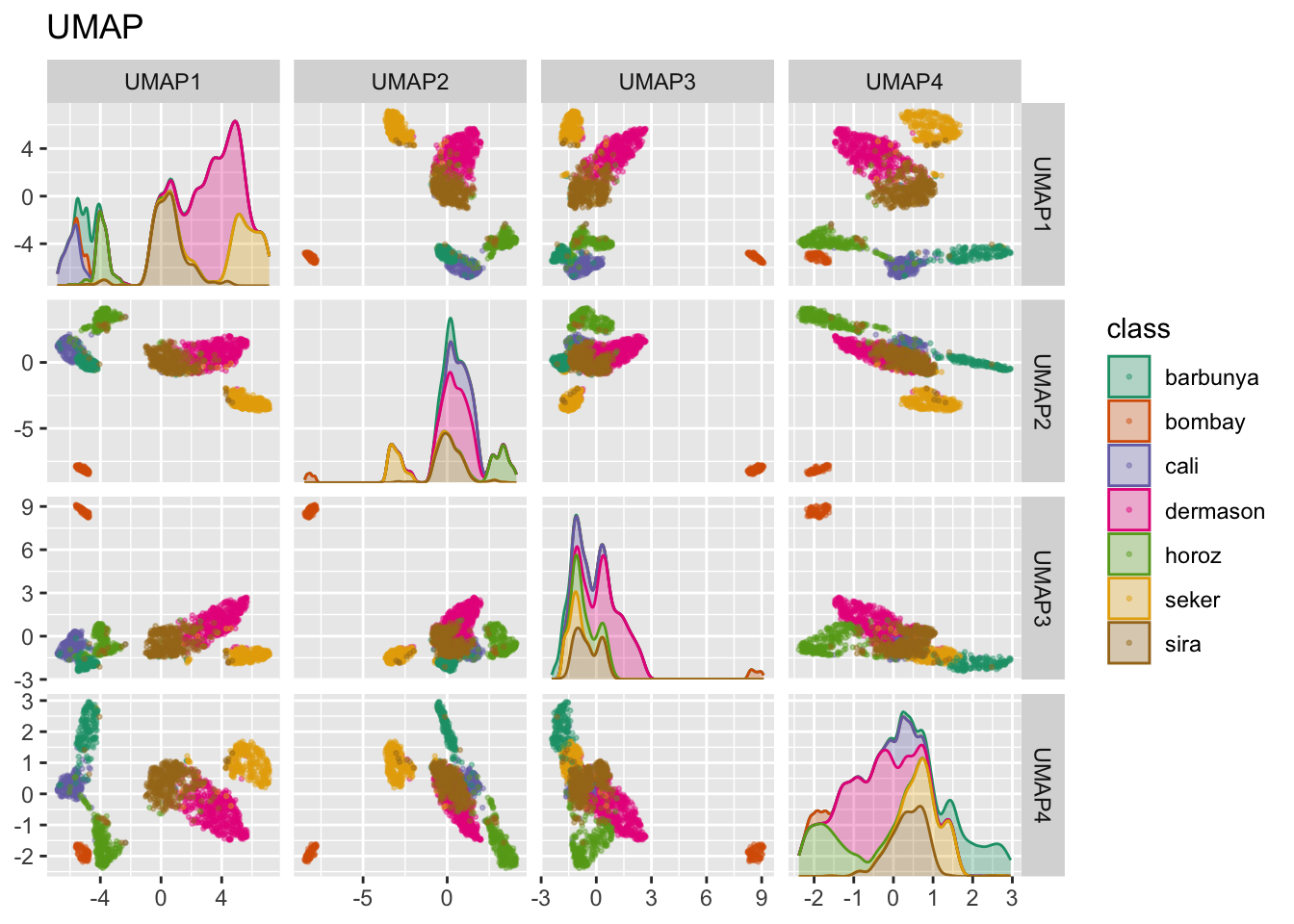
Widać wyraźne przestrzenie pomiędzy skupieniami, choć te dalej mogą być heterogeniczne.
Istnieje też odmiana UMAP nadzorowana, która zamiast używać podobieństwa do określenia skupień w wielowymiarowej przestrzeni, używa etykiet.
bean_rec_trained %>%
step_umap(all_numeric_predictors(), outcome = "class", num_comp = 4) %>%
plot_validation_results() +
ggtitle("UMAP (supervised)")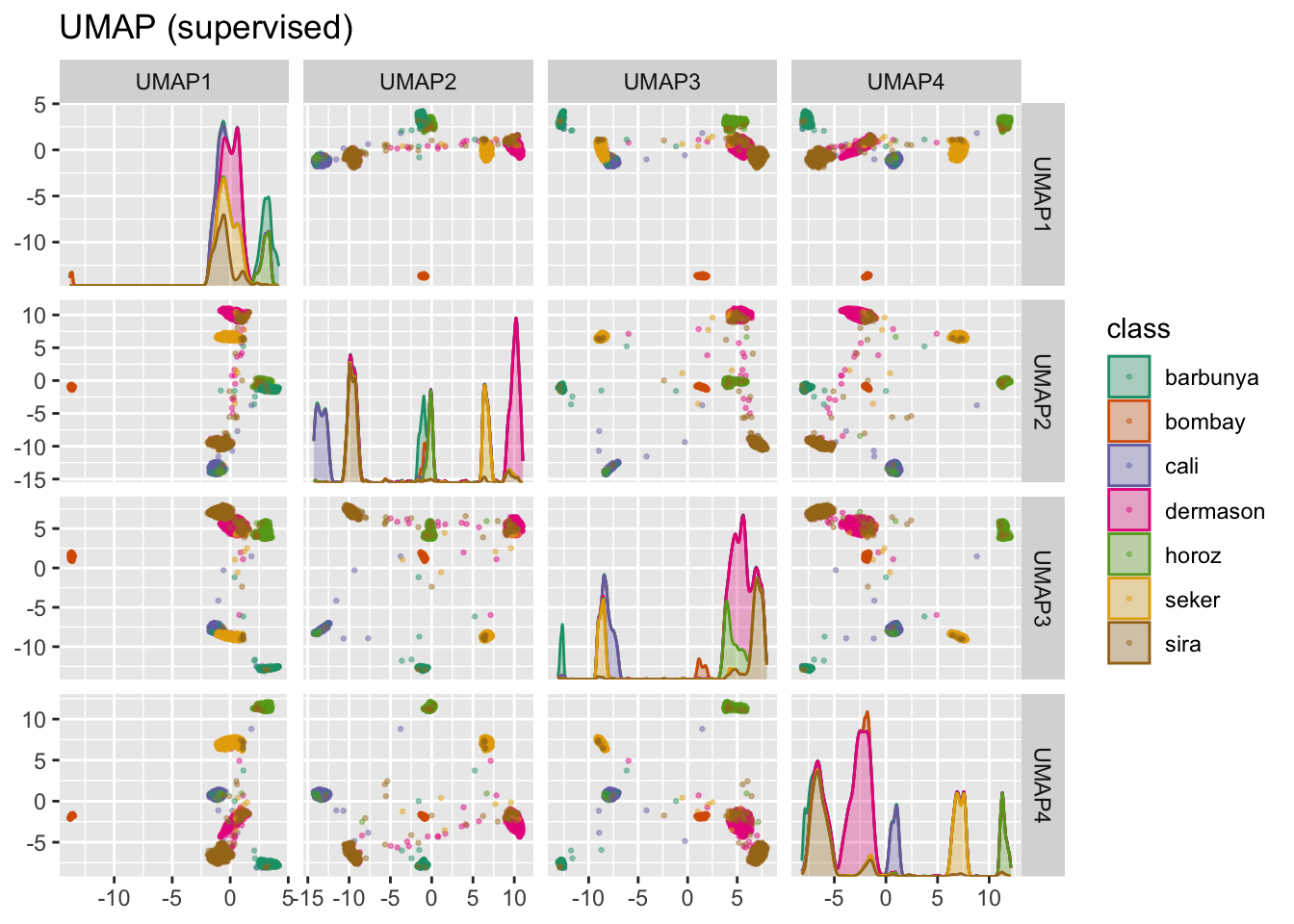
UMAP jest wydajną metodą redukcji przestrzeni cech. Jednak może być bardzo wrażliwa na dostrajanie parametrów (np. liczba sąsiadów). Z tego powodu pomocne byłoby eksperymentowanie z kilkoma parametrami, aby ocenić, jak wiarygodne są wyniki dla tych danych.
Zarówno metoda PLS jak i UMAP są warte zbadania w połączeniu z różnymi modelami. Zbadajmy wiele różnych modeli z tymi technikami redukcji wymiarowości (wraz z brakiem jakiejkolwiek transformacji): jednowarstwowa sieć neuronowa, bagged trees, elastyczna analiza dyskryminacyjna (FDA), naiwny Bayes i regularyzowana analiza dyskryminacyjna (RDA).
Stworzymy serię specyfikacji modeli, a następnie użyjemy zestawu przepływów pracy do dostrojenia modeli w poniższym kodzie. Zauważ, że parametry modelu są dostrajane w połączeniu z parametrami receptury (np. rozmiar zredukowanego wymiaru, parametry UMAP).
library(baguette)
library(discrim)
mlp_spec <-
mlp(hidden_units = tune(), penalty = tune(), epochs = tune()) %>%
set_engine('nnet') %>%
set_mode('classification')
bagging_spec <-
bag_tree() %>%
set_engine('rpart') %>%
set_mode('classification')
fda_spec <-
discrim_flexible(
prod_degree = tune()
) %>%
set_engine('earth')
rda_spec <-
discrim_regularized(frac_common_cov = tune(), frac_identity = tune()) %>%
set_engine('klaR')
bayes_spec <-
naive_Bayes() %>%
set_engine('klaR')Potrzebujemy również receptur dla metod redukcji wymiarowości, które będziemy testować. Zacznijmy od bazowego przepisu bean_rec, a następnie rozszerzmy go o różne kroki redukcji wymiarowości:
bean_rec <-
recipe(class ~ ., data = bean_train) %>%
step_zv(all_numeric_predictors()) %>%
step_orderNorm(all_numeric_predictors()) %>%
step_normalize(all_numeric_predictors())
pls_rec <-
bean_rec %>%
step_pls(all_numeric_predictors(), outcome = "class", num_comp = tune())
umap_rec <-
bean_rec %>%
step_umap(
all_numeric_predictors(),
outcome = "class",
num_comp = tune(),
neighbors = tune(),
min_dist = tune()
)Po raz kolejny pakiet workflowsets bierze reguły wstępnego przetwarzani oraz modele i krzyżuje je. Opcja kontrolna parallel_over jest ustawiona tak, aby przetwarzanie równoległe mogło działać jednocześnie w różnych kombinacjach dostrajania parametrów. Funkcja workflow_map() stosuje przeszukiwanie siatki w celu optymalizacji parametrów modelu/przetwarzania wstępnego (jeśli istnieją) w 10 kombinacjach parametrów. Obszar pod krzywą ROC jest szacowany na zbiorze walidacyjnym.
ctrl <- control_grid(parallel_over = "resamples")
bean_res <-
workflow_set(
preproc = list(basic = class ~., pls = pls_rec, umap = umap_rec),
models = list(bayes = bayes_spec, fda = fda_spec,
rda = rda_spec, bag = bagging_spec,
mlp = mlp_spec)
) %>%
workflow_map(
verbose = TRUE,
seed = 1603,
resamples = bean_val,
grid = 10,
metrics = metric_set(roc_auc),
control = ctrl
)bean_res <- readRDS("models/bean_res.rds")
autoplot(
bean_res,
rank_metric = "roc_auc", # <- how to order models
metric = "roc_auc", # <- which metric to visualize
select_best = TRUE # <- one point per workflow
) +
geom_text(aes(y = mean - 0.02, label = wflow_id), angle = 90, hjust = 0.1) +
lims(y = c(0.9, 1))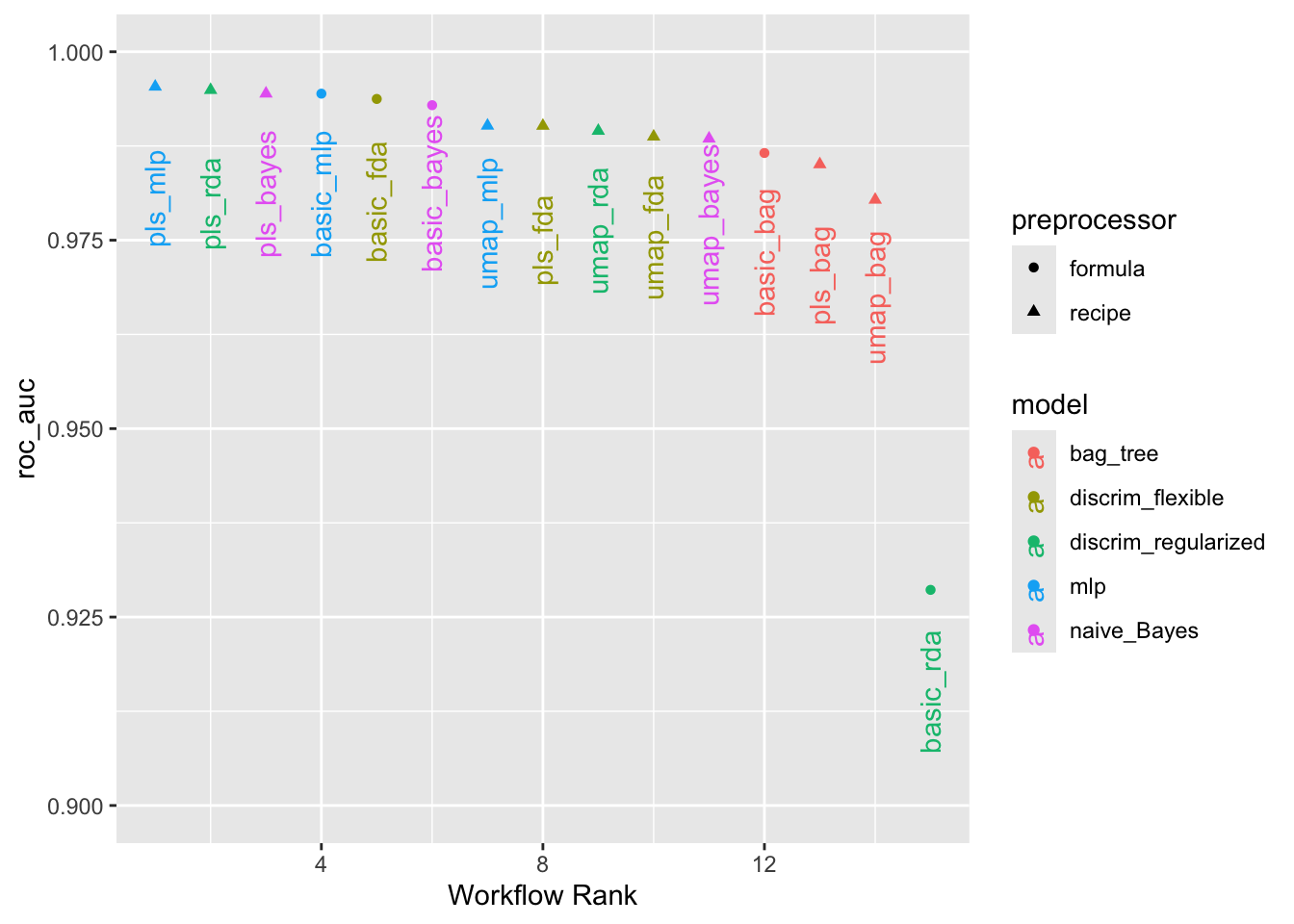
Możemy uszeregować modele według ich oszacowań obszaru pod krzywą ROC w zbiorze walidacyjnym:
# A tibble: 5 × 4
rank mean model method
<int> <dbl> <chr> <chr>
1 1 0.995 mlp pls
2 2 0.995 discrim_regularized pls
3 3 0.994 naive_Bayes pls
4 4 0.994 mlp basic
5 5 0.994 discrim_flexible basic Z tych wyników jasno wynika, że większość modeli daje bardzo dobre wyniki; niewiele jest tu złych wyborów. Dla demonstracji użyjemy modelu RDA z cechami PLS jako modelu końcowego.
# A tibble: 1 × 4
.metric .estimator .estimate .config
<chr> <chr> <dbl> <chr>
1 roc_auc hand_till 0.995 Preprocessor1_Model1

